
es-tr
-
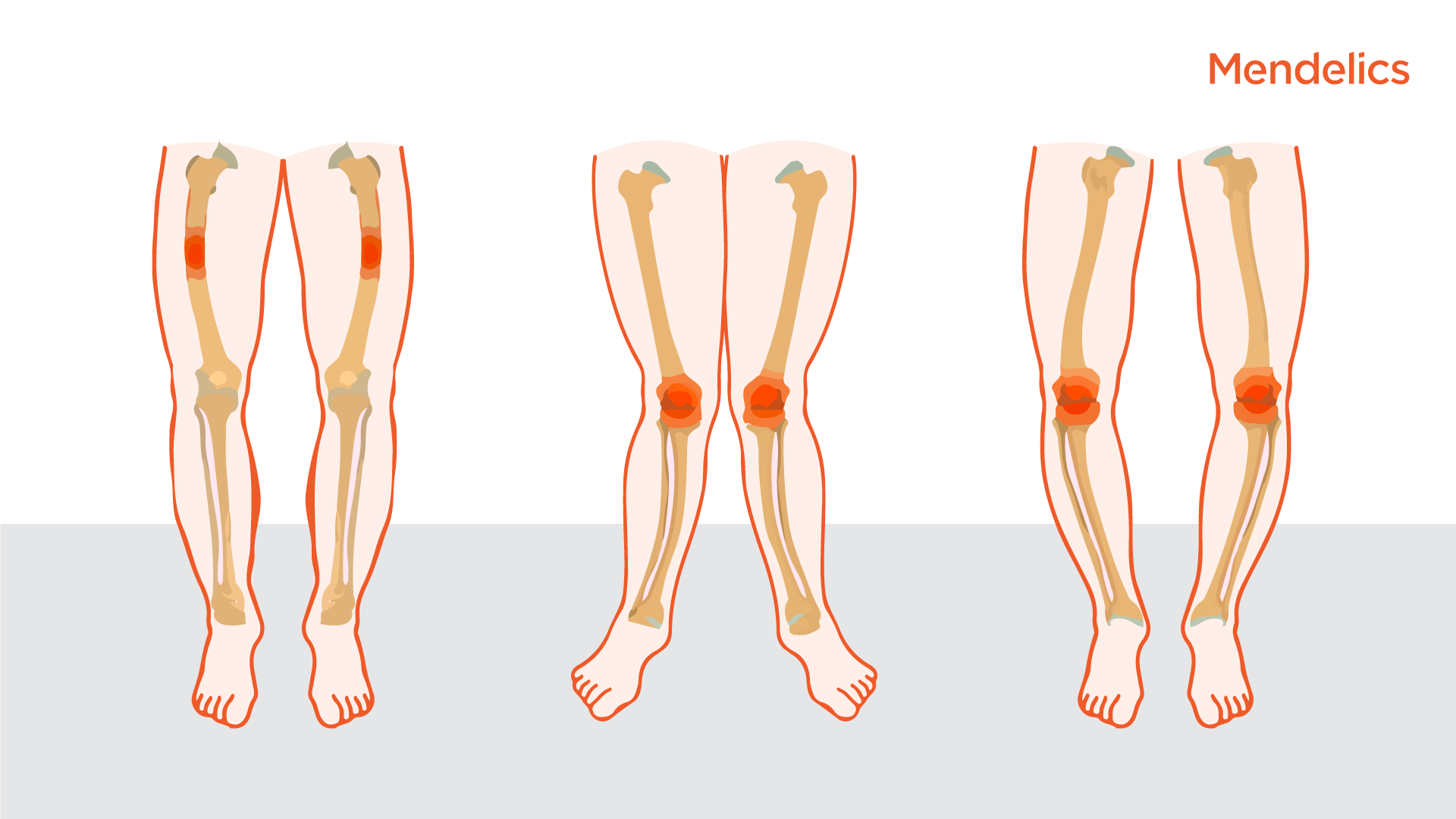
Raquitismo hipofosfatémico: genética, diagnóstico y tratamiento
El raquitismo hipofosfatémico es una enfermedad rara y progresiva causada por una insuficiencia renal, lo que hace que los huesos sean dolorosamente blandos.
-

Día Internacional de la Lucha contra el Cáncer Infantil
Cada año, unos 400 mil niños y adolescentes (hasta los 19 años) son diagnosticados de algún tipo de cáncer. Estos casos, llamados de cáncer infantil, ya representan la principal causa de muerte de este grupo de edad en países de Latinoamérica y en muchos países desarrollados. Se estima que por lo menos 29 mil niños…