
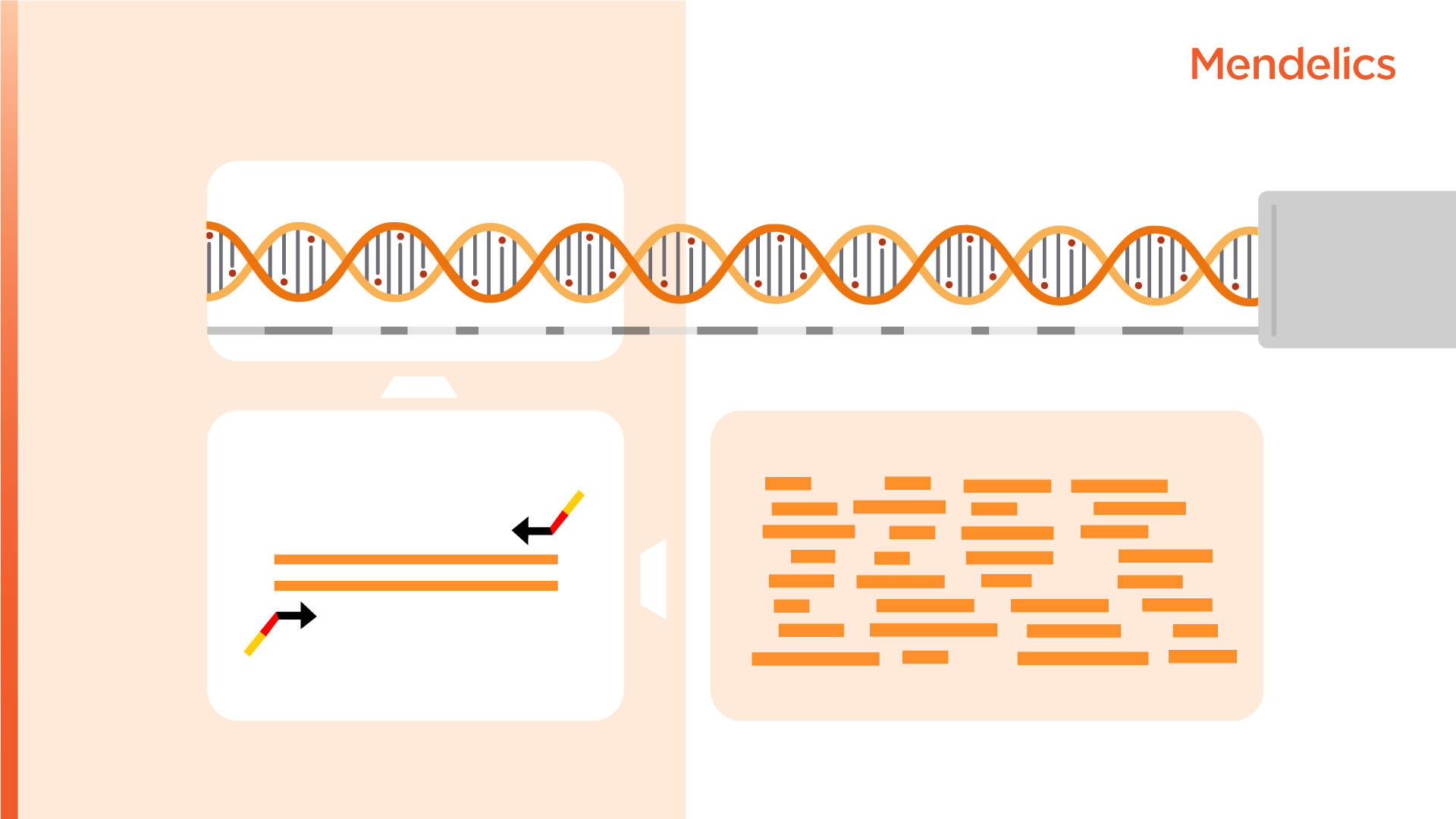
¿Qué es NGS Targeted Sequencing?
Las técnicas de secuenciación de nueva generación (del inglés Next Generation Sequencing, NGS) desarrolladas en las últimas décadas permitieron la secuenciación de genomas completos de varios individuos simultáneamente, reduciendo significativamente el costo y tiempo de los análisis. La tecnología de secuenciación dirigida NGS permite seleccionar regiones específicas del genoma (regiones objetivo que compondrán una biblioteca), haciendo que el análisis sea aún más rápido y menos costoso.
Las principales técnicas utilizadas en la actualidad son la secuenciación de amplicones (secuenciación por amplificación) y la secuenciación de captura basada en hibridación.
Comprenda más sobre NGS en este artículo.
Secuenciación por amplificación
En esta técnica, las regiones de ADN que se analizarán se seleccionan utilizando cebadores específicos. Estos cebadores son pequeñas secuencias de bases que se complementan entre sí con las regiones de ADN de interés. A continuación, los fragmentos seleccionados se amplifican mediante PCR para aumentar el número de copias en la biblioteca y facilitar la detección. A estas copias las llamamos amplicones.
Los cebadores utilizados, además de seleccionar las regiones del ADN a analizar, también añaden adaptadores a los extremos de los amplicones. Estos adaptadores contienen los índices (secuencias cortas de bases que identifican cada muestra), y secuencias complementarias a los cebadores de la flowcell (placa donde tiene lugar la secuenciación), utilizados en la secuenciación (Figura 1).

A pesar de ser una técnica más sencilla y económica, tiene algunas desventajas:
- Cada región de interés requiere de un par de cebadores específicos, lo que limita el número de regiones que pueden analizarse simultáneamente mediante este tipo de técnica (1,2).
- El cebador se une a la cadena de ADN a una temperatura específica que puede variar para cada par de cebadores. Por lo tanto, algunos amplicones se multiplicarán más fácil y eficientemente que otros, resultando en una cobertura desigual (número de veces que se representa una región) de las diferentes regiones de interés (1,2).
- Los cebadores deben ser complementarios a la cadena de ADN donde se unirán. La presencia de variaciones (mutaciones) puede prevenir la amplificación (dropout). Los eventos de dropout pueden resultar tanto en la pérdida de cobertura, cuando no se amplifica la copia materna ni paterna, como en el genotipado erróneo de homocigotos (individuos con dos copias iguales de un punto de variación), cuando solo se secuencia una copia, disminuyendo la precisión de la técnica (1,2).
Secuenciación por Hibridación y Captura
En esta técnica, el ADN se fragmenta en piezas más pequeñas que se sobreponen. Los fragmentos que contienen las regiones de interés son unidos a los adaptadores que tienen los índices de identificación de la muestra y las secuencias complementarias a los cebadores de la flowcell. Estos fragmentos se hibridan (enlazan) con sondas que tienen una molécula de biotina unida y se utilizan para capturar los fragmentos de interés.
La secuenciación por hibridación y captura no utiliza amplicones (Figura 2) para enriquecer la biblioteca con las regiones de interés, por lo que se resuelven los problemas encontrados en los análisis de secuenciación por amplificación.
- La secuenciación por hibridación y captura es capaz de captar todas las regiones de interés (no se produce ningún dropout), presentando una mayor sensibilidad y precisión (1,2).
- Presenta una cobertura más homogénea de los fragmentos secuenciados, además de permitir la secuenciación de un número prácticamente infinito de regiones de interés (1,2,3)

Así, aunque la técnica de Secuenciación por Hibridación y Captura es más compleja y puede tener un costo más elevado, permite un análisis más sensible y preciso de los genes de interés. Por esta razón, Mendelics utiliza kits para preparar bibliotecas de secuenciación de captura e hibridación para análisis y diagnóstico genético.
Referencias
- G. Matthijs et al., “Guidelines for diagnostic next-generation sequencing”, European Journal of Human Genetics, vol. 24, no. 1, pp. 2–5, Oct. 2015, doi: 10.1038/ejhg.2015.226.
- E. Samorodnitsky et al., “Evaluation of hybridization capture versus Amplicon‐Based methods for Whole Exome sequencing”, Human Mutation, vol. 36, no. 9, pp. 903–914, Jul. 2015, doi: 10.1002/humu.22825.
- S. S. Hung et al., “Assessment of capture and amplicon-based approaches for the development of a targeted next-generation sequencing pipeline to personalize lymphoma management,” The Journal of Molecular Diagnostics, vol. 20, no. 2, pp. 203–214, Mar. 2018, doi: 10.1016/j.jmoldx.2017.11.010.
Posts relacionados
 Mendelics tiene acreditaciones CAP, Inmetro y PALC
Mendelics tiene acreditaciones CAP, Inmetro y PALC
 Bancos de datos genéticos, enfermedades raras y enfermedades comunes
Bancos de datos genéticos, enfermedades raras y enfermedades comunes
 Bancos de datos genéticos – ClinVar
Bancos de datos genéticos – ClinVar
 CRISPR/Cas9: edición de adn y tratamiento de enfermedades
CRISPR/Cas9: edición de adn y tratamiento de enfermedades
 Conozca el panel de enfermedades tratables
Conozca el panel de enfermedades tratables
 Entienda sobre secuenciación de nueva generación (NGS)
Entienda sobre secuenciación de nueva generación (NGS)











Deja un comentario